| Hand abrasion | ||||
| Approximate days since injury | ||||
| 0 | 2 | 17 | 30 | |
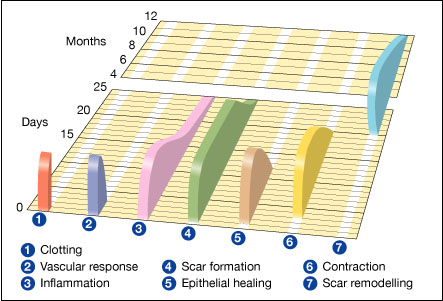
Wound healing, or cicatrisation, is an intricate process in which the skin (or another organ-tissue) repairs itself after injury.[1] In normal skin, the epidermis (outermost layer) and dermis (inner or deeper layer) exists in a steady-state equilibrium, forming a protective barrier against the external environment. Once the protective barrier is broken, the normal (physiologic) process of wound healing is immediately set in motion. The classic model of wound healing is divided into three or four sequential, yet overlapping,[2] phases: (1) hemostasis (not considered a phase by some authors), (2) inflammatory, (3) proliferative and (4) remodeling.[3] Upon injury to the skin, a set of complex biochemical events takes place in a closely orchestrated cascade to repair the damage.[2] Within minutes post-injury, platelets (thrombocytes) aggregate at the injury site to form a fibrin clot. This clot acts to control active bleeding (hemostasis).
In the inflammatory phase, bacteria and debris are phagocytosed and removed, and factors are released that cause the migration and division of cells involved in the proliferative phase.
The proliferative phase is characterized by angiogenesis, collagen deposition, granulation tissue formation, epithelialization, and wound contraction.[4] In angiogenesis, new blood vessels are formed by vascular endothelial cells.[5] In fibroplasia and granulation tissue formation, fibroblasts grow and form a new, provisional extracellular matrix (ECM) by excreting collagen and fibronectin.[4] Concurrently, re-epithelialization of the epidermis occurs, in which epithelial cells proliferate and 'crawl' atop the wound bed, providing cover for the new tissue.[6]
In contraction, the wound is made smaller by the action of myofibroblasts, which establish a grip on the wound edges and contract themselves using a mechanism similar to that in smooth muscle cells. When the cells' roles are close to complete, unneeded cells undergo apoptosis.[4]
In the maturation and remodeling phase, collagen is remodeled and realigned along tension lines and cells that are no longer needed are removed by apoptosis.
However, this process is not only complex but fragile, and susceptible to interruption or failure leading to the formation of non-healing chronic wounds. Factors which may contribute to this include diabetes, venous or arterial disease, old age, and infection.[7]
Contents |
As mentioned above, wound healing is classically divided into hemostasis, inflammation, proliferation, and remodeling. Although a useful construct, this model employs considerable overlapping among individual phases. Recently, a complementary model has been described,[1] such that the many elements of wound healing are more-clearly delineated. The importance of this new model becomes more apparent through its utility in the fields of regenerative medicine and tissue engineering (see Research and development section below). In this construct, the process of wound healing is divided into major two phases: early phase and cellular phase:[1]
The early phase, which begins immediately following skin injury, involves cascading molecular and cellular events leading to hemostasis and formation of an early, makeshift extracellular matrix—providing structural support for cellular attachment and subsequent cellular proliferation.
The cellular phase follows the early phase, and involves several types of cells working together to mount an inflammatory response, synthesize granulation tissue, and restore the epithelial layer.[1] Subdivisions of the cellular phase are: [1] Macrophages and inflammatory components (within 1–2 days), [2] Epithelial-mesenchymal interaction: re-epithelialization (phenotype change within hours, migration begins on day 1 or 2, [3] Fibroblasts and myofibroblasts: progressive alignment, collagen production, and matrix contraction (between day 4 day 14), [4] Endothelial cells and angiogenesis (begins on day 4), [5] Dermal matrix: elements of fabrication (begins on day 4, lasting 2 weeks) and alteration/remodeling (begins after week 2, lasting weeks to months—depending on wound size.).[1]
Just before the inflammatory phase is initiated, the clotting cascade takes place in order to obtain hemostasis, or stop blood loss by way of a fibrin clot. Thereafter, various soluble factors (including chemokines and cytokines) are released to attract cells that phagocytise debris, bacteria, and damaged tissue, in addition to releasing signaling molecules that initiate the proliferative phase of wound healing.
When tissue is first wounded, blood comes in contact with collagen, triggering blood platelets to begin secreting inflammatory factors.[10] Platelets also express glycoproteins on their cell membranes that allow them to stick to one another and to aggregate, forming a mass.[4]
Fibrin and fibronectin cross-link together and form a plug that traps proteins and particles and prevents further blood loss.[11] This fibrin-fibronectin plug is also the main structural support for the wound until collagen is deposited.[4] Migratory cells use this plug as a matrix to crawl across, and platelets adhere to it and secrete factors.[4] The clot is eventually lysed and replaced with granulation tissue and then later with collagen.
Platelets, the cells present in the highest numbers shortly after a wound occurs, release a number of things into the blood, including ECM proteins and cytokines, including growth factors.[10] Growth factors stimulate cells to speed their rate of division. Platelets also release other proinflammatory factors like serotonin, bradykinin, prostaglandins, prostacyclins, thromboxane, and histamine,[2] which serve a number of purposes, including to increase cell proliferation and migration to the area and to cause blood vessels to become dilated and porous.
Immediately after a blood vessel is breached, ruptured cell membranes release inflammatory factors like thromboxanes and prostaglandins that cause the vessel to spasm to prevent blood loss and to collect inflammatory cells and factors in the area.[2] This vasoconstriction lasts five to ten minutes and is followed by vasodilation, a widening of blood vessels, which peaks at about 20 minutes post-wounding.[2] Vasodilation is the result of factors released by platelets and other cells. The main factor involved in causing vasodilation is histamine.[2][10] Histamine also causes blood vessels to become porous, allowing the tissue to become edematous because proteins from the bloodstream leak into the extravascular space, which increases its osmolar load and draws water into the area.[2] Increased porosity of blood vessels also facilitates the entry of inflammatory cells like leukocytes into the wound site from the bloodstream.[12][13]
Within an hour of wounding, polymorphonuclear neutrophils (PMNs) arrive at the wound site and become the predominant cells in the wound for the first two days after the injury occurs, with especially high numbers on the second day.[14] They are attracted to the site by fibronectin, growth factors, and substances such as kinins. Neutrophils phagocytise debris and bacteria and also kill bacteria by releasing free radicals in what is called a 'respiratory burst'.[15][16] They also cleanse the wound by secreting proteases that break down damaged tissue. Neutrophils usually undergo apoptosis once they have completed their tasks and are engulfed and degraded by macrophages.[17]
Other leukocytes to enter the area include helper T cells, which secrete cytokines to cause more T cells to divide and to increase inflammation and enhance vasodilation and vessel permeability.[12][18] T cells also increase the activity of macrophages.[12]
Macrophages are essential for wound healing.[14] They replace PMNs as the predominant cells in the wound by two days after injury.[19] Attracted to the wound site by growth factors released by platelets and other cells, monocytes from the bloodstream enter the area through blood vessel walls.[20] Numbers of monocytes in the wound peak one to one and a half days after the injury occurs.[18] Once they are in the wound site, monocytes mature into macrophages. The spleen contains half the body's monocytes in reserve ready to be deployed to injured tissue.[21][22]
The macrophage's main role is to phagocytize bacteria and damaged tissue,[14] and they also debride damaged tissue by releasing proteases.[23] Macrophages also secrete a number of factors such as growth factors and other cytokines, especially during the third and fourth post-wounding days. These factors attract cells involved in the proliferation stage of healing to the area.,[10] although they may restrain the contraction phase.[24] Macrophages are stimulated by the low oxygen content of their surroundings to produce factors that induce and speed angiogenesis.[15] and they also stimulate cells that reepithelialize the wound, create granulation tissue, and lay down a new extracellular matrix.[25][26] By secreting these factors, macrophages contribute to pushing the wound healing process into the next phase.
As inflammation dies down, fewer inflammatory factors are secreted, existing ones are broken down, and numbers of neutrophils and macrophages are reduced at the wound site.[14] These changes indicate that the inflammatory phase is ending and the proliferative phase is underway.[14] In vitro evidence, obtained using the dermal equivalent model, suggests that the presence of macrophages actually delays wound contraction and thus the disappearance of macrophages from the wound may be essential for subsequent phases to occur.[24]
Because inflammation plays roles in fighting infection, clearing debris and inducing the proliferation phase, it is a necessary part of healing. However, inflammation can lead to tissue damage if it lasts too long.[4] Thus the reduction of inflammation is frequently a goal in therapeutic settings. Inflammation lasts as long as there is debris in the wound. Thus the presence of dirt or other objects can extend the inflammatory phase for too long, leading to a chronic wound.
About two or three days after the wound occurs, fibroblasts begin to enter the wound site, marking the onset of the proliferative phase even before the inflammatory phase has ended.[27] As in the other phases of wound healing, steps in the proliferative phase do not occur in a series but rather partially overlap in time.
Also called neovascularization, the process of angiogenesis occurs concurrently with fibroblast proliferation when endothelial cells migrate to the area of the wound.[28] Because the activity of fibroblasts and epithelial cells requires oxygen and nutrients, angiogenesis is imperative for other stages in wound healing, like epidermal and fibroblast migration. The tissue in which angiogenesis has occurred typically looks red (is erythematous) due to the presence of capillaries.[28]
Stem cells of endothelial cells, originating from parts of uninjured blood vessels, develop pseudopodia and push through the ECM into the wound site to establish new blood vessels.[15]
Endothelial cells are attracted to the wound area by fibronectin found on the fibrin scab and chemotactically by angiogenic factors released by other cells,[29] e.g. from macrophages and platelets when in a low-oxygen environment. Endothelial growth and proliferation is also directly stimulated by hypoxia, and presence of lactic acid in the wound.[27]
To migrate, endothelial cells need collagenases and plasminogen activator to degrade the clot and part of the ECM.[2][14] Zinc-dependent metalloproteinases digest basement membrane and ECM to allow cell migration, proliferation and angiogenesis.[30]
When macrophages and other growth factor-producing cells are no longer in a hypoxic, lactic acid-filled environment, they stop producing angiogenic factors.[15] Thus, when tissue is adequately perfused, migration and proliferation of endothelial cells is reduced. Eventually blood vessels that are no longer needed die by apoptosis.[29]
Simultaneously with angiogenesis, fibroblasts begin accumulating in the wound site. Fibroblasts begin entering the wound site two to five days after wounding as the inflammatory phase is ending, and their numbers peak at one to two weeks post-wounding.[14] By the end of the first week, fibroblasts are the main cells in the wound[2] Fibroplasia ends two to four weeks after wounding.
In the first two or three days after injury, fibroblasts mainly migrate and proliferate, while later, they are the main cells that lay down the collagen matrix in the wound site.[2] Origins of these fibroblasts are thought to be from the adjacent uninjured cutaneous tissue (although new evidence suggests that some are derived from blood-borne, circulating adult stem cells/precursors).[31] Initially fibroblasts utilize the fibrin cross-linking fibers (well-formed by the end of the inflammatory phase) to migrate across the wound, subsequently adhering to fibronectin.[29] Fibroblasts then deposit ground substance into the wound bed, and later collagen, which they can adhere to for migration.[10]
Granulation tissue functions as rudimentary tissue, and begins to appear in the wound already during the inflammatory phase, two to five days post wounding, and continues growing until the wound bed is covered. Granulation tissue consists of new blood vessels, fibroblasts, inflammatory cells, endothelial cells, myofibroblasts, and the components of a new, provisional extracellular matrix (ECM). The provisional ECM is different in composition from the ECM in normal tissue and its components originate from fibroblasts.[25] Such components include fibronectin, collagen, glycosaminoglycans, elastin, glycoproteins and proteoglycans.[29] Its main components are fibronectin and hyaluronan, which create a very hydrated matrix and facilitate cell migration.[20] Later this provisional matrix is replaced with an ECM that more closely resembles that found in non-injured tissue.
Growth factors (PDGF, TGF-β) and fibronectin encourage proliferation, migration to the wound bed, and production of ECM molecules by fibroblasts. Fibroblasts also secrete growth factors that attract epithelial cells to the wound site. Hypoxia also contributes to fibroblast proliferation and excretion of growth factors, though too little oxygen will inhibit their growth and deposition of ECM components, and can lead to excessive, fibrotic scarring.
One of fibroblasts' most important duties is the production of collagen.[28]
Collagen deposition is important because it increases the strength of the wound; before it is laid down, the only thing holding the wound closed is the fibrin-fibronectin clot, which does not provide much resistance to traumatic injury.[15] Also, cells involved in inflammation, angiogenesis, and connective tissue construction attach to, grow and differentiate on the collagen matrix laid down by fibroblasts.[32]
Type III collagen and fibronectin are generally beginning to be produced in appreciable amounts at somewhere between approximately 10 hours[33] and 3 days,[29] depending mainly on wound size. Their deposition peaks at one to three weeks.[25] They are the predominating tensile substances until the later phase of maturation, in which they are replaced by the stronger type I collagen.
Even as fibroblasts are producing new collagen, collagenases and other factors degrade it. Shortly after wounding, synthesis exceeds degradation so collagen levels in the wound rise, but later production and degradation become equal so there is no net collagen gain.[15] This homeostasis signals the onset of the later maturation phase. Granulation gradually ceases and fibroblasts decrease in number in the wound once their work is done.[34] At the end of the granulation phase, fibroblasts begin to commit apoptosis, converting granulation tissue from an environment rich in cells to one that consists mainly of collagen.[2]
The formation of granulation tissue in an open wound allows the reepithelialization phase to take place, as epithelial cells migrate across the new tissue to form a barrier between the wound and the environment.[29] Basal keratinocytes from the wound edges and dermal appendages such as hair follicles, sweat glands and sebacious (oil) glands are the main cells responsible for the epithelialization phase of wound healing.[34] They advance in a sheet across the wound site and proliferate at its edges, ceasing movement when they meet in the middle.
Keratinocytes migrate without first proliferating.[35] Migration can begin as early as a few hours after wounding. However, epithelial cells require viable tissue to migrate across, so if the wound is deep it must first be filled with granulation tissue.[36] Thus the time of onset of migration is variable and may occur about one day after wounding.[37] Cells on the wound margins proliferate on the second and third day post-wounding in order to provide more cells for migration.[25]
If the basement membrane is not breached, epithelial cells are replaced within three days by division and upward migration of cells in the stratum basale in the same fashion that occurs in uninjured skin.[29] However, if the basement membrane is ruined at the wound site, reepithelization must occur from the wound margins and from skin appendages such as hair follicles and sweat and oil glands that enter the dermis that are lined with viable keratinocytes.[25] If the wound is very deep, skin appendages may also be ruined and migration can only occur from wound edges.[36]
Migration of keratinocytes over the wound site is stimulated by lack of contact inhibition and by chemicals such as nitric oxide.[38] Before they begin to migrate, cells must dissolve their desmosomes and hemidesmosomes, which normally anchor the cells by intermediate filaments in their cytoskeleton to other cells and to the ECM.[18] Transmembrane receptor proteins called integrins, which are made of glycoproteins and normally anchor the cell to the basement membrane by its cytoskeleton, are released from the cell's intermediate filaments and relocate to actin filaments to serve as attachments to the ECM for pseudopodia during migration.[18] Thus keratinocytes detach from the basement membrane and are able to enter the wound bed.[27]
Before they begin migrating, keratinocytes change shape, becoming longer and flatter and extending cellular processes like lamellipodia and wide processes that look like ruffles.[20] Actin filaments and pseudopodia form.[27] During migration, integrins on the pseudopod attach to the ECM, and the actin filaments in the projection pull the cell along.[18] The interaction with molecules in the ECM through integrins further promotes the formation of actin filaments, lamellipodia, and filopodia.[18]
Epithelial cells climb over one another in order to migrate.[34] This growing sheet of epithelial cells is often called the epithelial tongue.[35] The first cells to attach to the basement membrane form the stratum basale. These basal cells continue to migrate across the wound bed, and epithelial cells above them slide along as well.[35] The more quickly this migration occurs, the less of a scar there will be.[39]
Fibrin, collagen, and fibronectin in the ECM may further signal cells to divide and migrate. Like fibroblasts, migrating keratinocytes use the fibronectin cross-linked with fibrin that was deposited in inflammation as an attachment site to crawl across.[20][23][34]
As keratinocytes migrate, they move over granulation tissue but underneath the scab (if one was formed), separating it from the underlying tissue.[34][37] Epithelial cells have the ability to phagocytize debris such as dead tissue and bacterial matter that would otherwise obstruct their path. Because they must dissolve any scab that forms, keratinocyte migration is best enhanced by a moist environment, since a dry one leads to formation of a bigger, tougher scab.[23][29][34][40] To make their way along the tissue, keratinocytes must dissolve the clot, debris, and parts of the ECM in order to get through.[37][41] They secrete plasminogen activator, which activates plasminogen, turning it into plasmin to dissolve the scab. Cells can only migrate over living tissue,[34] so they must excrete collagenases and proteases like matrix metalloproteinases (MMPs) to dissolve damaged parts of the ECM in their way, particularly at the front of the migrating sheet.[37] Keratinocytes also dissolve the basement membrane, using instead the new ECM laid down by fibroblasts to crawl across.[18]
As keratinocytes continue migrating, new epithelial cells must be formed at the wound edges to replace them and to provide more cells for the advancing sheet.[23] Proliferation behind migrating keratinocytes normally begins a few days after wounding[36] and occurs at a rate that is 17 times higher in this stage of epithelialization than in normal tissues.[23] Until the entire wound area is resurfaced, the only epithelial cells to proliferate are at the wound edges.[35]
Growth factors, stimulated by integrins and MMPs, cause cells to proliferate at the wound edges. Keratinocytes themselves also produce and secrete factors, including growth factors and basement membrane proteins, which aid both in epithelialization and in other phases of healing.[42] Growth factors are also important for the innate immune defense of skin wounds by stimulation of the production of antimicrobial peptides and neutrophil chemotactic cytokines in keratinocytes.[43]
Keratinocytes continue migrating across the wound bed until cells from either side meet in the middle, at which point contact inhibition causes them to stop migrating.[20] When they have finished migrating, the keratinocytes secrete the proteins that form the new basement membrane.[20] Cells reverse the morphological changes they underwent in order to begin migrating; they reestablish desmosomes and hemidesmosomes and become anchored once again to the basement membrane.[18] Basal cells begin to divide and differentiate in the same manner as they do in normal skin to reestablish the strata found in reepithelialized skin.[20]
Contraction is a key phase of wound healing. If contraction continues for too long, it can lead to disfigurement and loss of function.[44] Thus there is a great interest in understanding the biology of wound contraction, which can be modelled in vitro using the collagen gel contraction assay or the dermal equivalent model.[24][45]
Contraction commences approximately a week after wounding, when fibroblasts have differentiated into myofibroblasts [46] In full thickness wounds, contraction peaks at 5 to 15 days post wounding.[29] Contraction can last for several weeks[36] and continues even after the wound is completely reepithelialized.[2] A large wound can become 40 to 80% smaller after contraction.[20][34] Wounds can contract at a speed of up to 0.75 mm per day, depending on how loose the tissue in the wounded area is.[29] Contraction usually does not occur symmetrically; rather most wounds have an 'axis of contraction' which allows for greater organization and alignment of cells with collagen.[46]
At first, contraction occurs without myofibroblast involvement.[47] Later, fibroblasts, stimulated by growth factors, differentiate into myofibroblasts. Myofibroblasts, which are similar to smooth muscle cells, are responsible for contraction.[47] Myofibroblasts contain the same kind of actin as that found in smooth muscle cells.[44]
Myofibroblasts are attracted by fibronectin and growth factors and they move along fibronectin linked to fibrin in the provisional ECM in order to reach the wound edges.[23] They form connections to the ECM at the wound edges, and they attach to each other and to the wound edges by desmosomes. Also, at an adhesion called the fibronexus, actin in the myofibroblast is linked across the cell membrane to molecules in the extracellular matrix like fibronectin and collagen.[47] Myofibroblasts have many such adhesions, which allow them to pull the ECM when they contract, reducing the wound size.[44] In this part of contraction, closure occurs more quickly than in the first, myofibroblast-independent part.[47]
As the actin in myofibroblasts contracts, the wound edges are pulled together. Fibroblasts lay down collagen to reinforce the wound as myofibroblasts contract[2] The contraction stage in proliferation ends as myofibroblasts stop contracting and commit apoptosis.[44] The breakdown of the provisional matrix leads to a decrease in hyaluronic acid and an increase in chondroitin sulfate, which gradually triggers fibroblasts to stop migrating and proliferating.[14] These events signal the onset of the maturation stage of wound healing.
When the levels of collagen production and degradation equalize, the maturation phase of tissue repair is said to have begun.[15] During maturation, type III collagen, which is prevalent during proliferation, is gradually degraded and the stronger type I collagen is laid down in its place.[12] Originally disorganized collagen fibers are rearranged, cross-linked, and aligned along tension lines.[20] The onset of the maturation phase may vary extensively, depending on the size of the wound and whether it was initially closed or left open,[25] ranging from approximately 3 days[33] to 3 weeks[48] The maturation phase can last for a year or longer, similarly depending on wound type.[25]
As the phase progresses, the tensile strength of the wound increases, with the strength approaching 50% that of normal tissue by three months after injury and ultimately becoming as much as 80% as strong as normal tissue.[25] Since activity at the wound site is reduced, the scar loses its red appearance as blood vessels that are no longer needed are removed by apoptosis.[15]
The phases of wound healing normally progress in a predictable, timely manner; if they do not, healing may progress inappropriately to either a chronic wound [4] such as a venous ulcer or pathological scarring such as a keloid scar.[49][50]
Up until a decade ago, the classic paradigm of wound healing, involving stem cells restricted to organ-specific lineages, had never been seriously challenged. Since then, the notion of adult stem cells having cellular plasticity or the ability to differentiate into non-lineage cells has emerged as an alternative explanation.[1] To be more specific, hematopoietic progenitor cells (that give rise to mature cells in the blood) may have the ability de-differentiate back into hematopoietic stem cells and/or transdifferentiate into non-lineage cells, such as fibroblasts.[31]
Multipotent adult stem cells have the capacity to be self-renewing and give rise to different cell types. Stem cells give rise to progenitor cells, which are cells that are not self-renewing, but can generate several types of cells. The extent of stem cell involvement in cutaneous (skin) wound healing is complex and not fully understood.
It is thought that the epidermis and dermis are reconstituted by mitotically active stem cells that reside at the apex of rete ridges (basal stem cells or BSC), the bulge of hair follicles (hair follicular stem cell or HFSC), and the papillary dermis (dermal stem cells).[1] Moreover, bone marrow may also contain stem cells that play a major role in cutaneous wound healing.[31]
In rare circumstances, such as extensive cutaneous injury, self-renewal subpopulations in the bone marrow are induced to participate in the healing process, whereby they give rise to collagen-secreting cells that seem to play a role during wound repair.[1] These two self-renewal subpopulations are (1) bone marrow-derived mesenchymal stem cells (MSC) and (2) hematopoietic stem cells (HSC). Bone marrow also harbors a progenitor subpopulation (endothelial progenitor cells or EPC) that, in the same type of setting, are mobilized to aid in the reconstruction of blood vessels.[31] Moreover, it thought that, extensive injury to skin also promotes the early trafficking of a unique subclass of leukocytes (circulating fibrocytes) to the injured region, where they perform various functions related to wound healing.[1]
There is a subtle distinction between ‘repair’ and ‘regeneration'.[1] An injury is an interruption of morphology and/or functionality of a given tissue. Repair refers to the physiologic adaptation of an organ after injury in an effort to re-establish continuity without regards to exact replacement of lost/damaged tissue. True tissue regeneration refers to the replacement of lost/damaged tissue with an ‘exact’ copy, such that both morphology and functionality are completely restored. Mammals do not regenerate spontaneously. In some instances, such as skin, ‘partial regeneration’ may be induced by the use of biodegradable (collagen-glycoaminoglycan) scaffolds. These scaffolds are structurally analogous to extracellular matrix (ECM) found in normal/un-injured dermis.[51] Interestingly, fundamental conditions required for tissue regeneration often oppose conditions that favor efficient wound repair, including inhibition of (1) platelet activation, (2) inflammatory response, and (3) wound contraction.[1] In addition to providing support for fibroblast and endothelial cell attachment, biodegradable scaffolds inhibit wound contraction, thereby allowing the healing process to proceed towards a more-regenerative/less-scarring pathway. Pharmaceutical agents have been investigated which may be able to turn off myofibroblast differentiation[52]
A new way of thinking derived from the notion that heparan sulfates are key player in tissue homeostasis: the process that makes the tissue replace dead cells by identical cells. In wound areas, tissue homeostasis is lost as the heparan sulfates are degraded preventing the replacement of dead cells by identical cells. Heparan sulfate analogues cannot be degraded by all know heparanases and glycanases and bind to the free heparin sulfate binding spots on the ECM, therefore preserving the normal tissue homeostasis and preventing scarring. [53] [54] [55]
involves epidermis and dermis without total penetration of dermis healing by process of epithelialization
(Delayed primary closure or secondary suture):
If the wound edges are not reapproximated immediately, delayed primary wound healing transpires. This type of healing may be desired in the case of contaminated wounds. By the fourth day, phagocytosis of contaminated tissues is well underway, and the processes of epithelization, collagen deposition, and maturation are occurring. Foreign materials are walled off by macrophages that may metamorphose into epithelioid cells, which are encircled by mononuclear leukocytes, forming granulomas. Usually the wound is closed surgically at this juncture, and if the "cleansing" of the wound is incomplete, chronic inflammation can ensue, resulting in prominent scarring.
Following are the main growth factors involved in wound healing:
| Growth factor | Abbreviation | Main origins | Effects |
|---|---|---|---|
| Epidermal growth factor | EGF |
|
|
| Transforming growth factor-α | TGF-α |
|
|
| Hepatocyte growth factor | HGF |
|
|
| Vascular endothelial growth factor | VEGF |
|
|
| Platelet derived growth factor | PDGF |
|
|
| Fibroblast growth factor 1 and 2 | FGF-1, -2 |
|
|
| Transforming growth factor-β | TGF-β |
|
|
| Keratinocyte growth factor | KGF |
|
|
| Unless else specified in boxes, then reference is:[56] | |||
|
||||||||||||||
|
||||||||
{kind=link}